Laboratory protocols
Dissection and Fixation
1. The animal is sacrificed by cervical dislocation and the head is cut off. The brain is extracted and placed into a dissecting dish filled with 0.1 M PBS.
2. The brain is divided and a coronally-oriented cut is then made at the posterior-most aspect of the interhemispheric fissure, allowing the caudal hippocampus to be visualized in cross-section.
3. The hippocampus is released from the overlying cortex using small strokes
4. Subsequently, the cortex is dissected away with small cuts by visualizing the interface between the corpus callosum and the SEZ.
Note: Simply cut along this interface staying on the callosal side to avoid damaging the SVZ.
5. Finally, the lateral wall is completely exposed by removing any overhanging cortex dorsally and the thalamus ventrally.
The wholemount is transferred (ventricle side up) from the dissection dish into a 24-well plate filled with 4% PFA with 0.1% Triton-X100 for an overnight fixation at 4°C.
Isolation and culture of neural stem cells from the adult mouse SEZ
1. The animal is sacrificed by cervical dislocation, the brain is extracted and placed into in a 60-mm Petri dish with ice-cold dissection medium (10mM of HEPES in HBSS 1×).
2. With the ventral side facing up, the brain is coronally transected using a surgical blade at the level of the optic chiasm. 3. The ventricle from the caudal side is opened up using forceps, allowing to visualize the ventricular face of the wall of the lateral ventricle. The SEZ is located just beneath the surface formed by a single layer of ependymal cells.
3. The perimeter of the SEZ is cut and afterwards, a thin layer beneath the surface is dissected, avoiding as much as possible the adjacent striatal tissue. The SEZs from 3 mice are pulled together and placed in a 15-ml conical tube containing 10 ml of dissection medium.
1. The pup is sacrificed by cutting the head off instantaneously, the brain is extracted and placed into in a 30-mm Petri dish with ice-cold dissection medium (10mM of HEPES in HBSS 1×).
2. The brain is divided and a coronally-oriented cut is made at the posterior-most aspect of the two hemispheres. The ventricle from the caudal side is opened up using forceps, allowing to visualize the ventricular face of the wall of the lateral ventricle.
3. Subsequently, the lateral wall is completely exposed and sub-dissected using fine forceps.
ChIP protocol for NS cells
Chromatin Preparation
- Aspirate the medium from a confluent 60mm plate and wash cells with PBS. Add 400μl Accutase and incubate 2-3min at 37oC. Collect the cells by adding medium and centrifuge at 1200rpm for 5min to pellet. Wash pellet once with PBS.
- Resuspend cells in 900μl PBS and add 100μl Formaldehyde 10% (CF = 1%) to fix the cells. Rotate at RT for 10min.
- Add 100μl glycine to a final concentration of 0.125M to stop fixation. Incubate 5min on ice and centrifuge at 2000rpm to pellet the cells. From that step work on ice.
- Remove supernatant and resuspend the pellet in cold PBS/PMSF and centrifuge at 2500rpm for 5min at 4o C. Repeat for two more times.
- Remove supernatant and resuspend pellet in Lysis buffer (1ml per 107 cells). Homogenize manually and incubate for 30min at 4oC rotating. Prepare aliquots of 300μl for sonication.
- Sonicate in Diagenode Bioraptor for 45 cycles (30sec ON/30sec OFF) settings: High.
- Centrifuge immediately at full speed for 15min. at 4oC. If necessary take the supernatant and centrifuge it again at full speed for 15min. Your supernatant has the sheared chromatin.
- Measure chromatin at nanodrop and dilute samples in Lysis buffer at final concentration of 1μg/μl. Make aliquots of 20μg. Aliquots can be kept at -800C until chromatin precipitation.
The size of the mitotic spindle during mitosis can give us information about the type of the division. During active neurogenesis Apical Radial Glia cells perform two types of divisions in order to self-renew or to differentiate, termed symmetric or asymmetric divisions respectively. For the evaluation of the division type we have to measure the volume of each spindle pole and calculate the difference between the two poles. This difference is the SSA index; high SSA index shows asymmetric divisions while a small number shows a symmetric division. To visualize the mitotic spindle in vivo, we perform immunofluorescence (IF) staining on cryosections of mouse embryos at E12.5 dpc with antibodies against α-tubulin and pericentrin, two main proteins of the spindle. To analyze, we take pictures in thin stacks 0,2μm and calculate the volume of each pole and subsequently the SSA index.
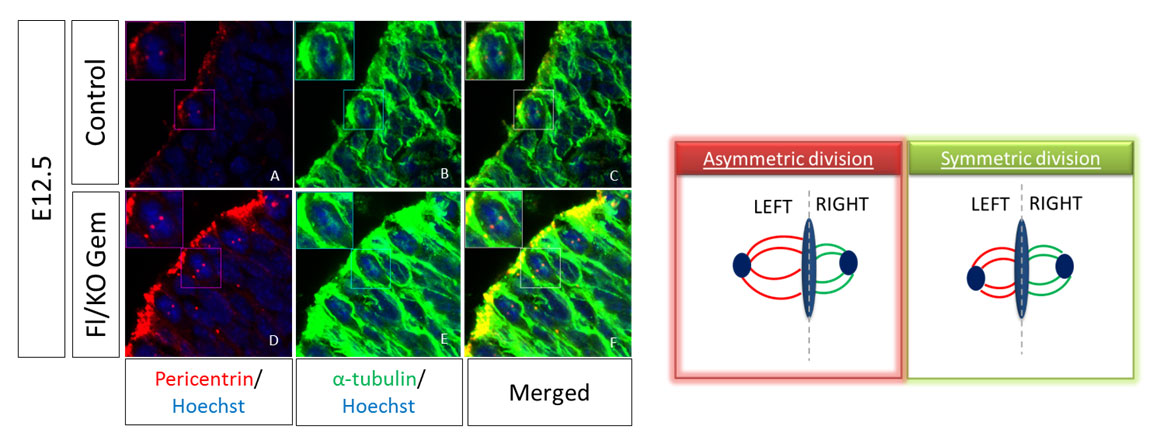 |
Read more: In vivo evaluation of Spindle Size Asymmetry (SSA)
 |